THE RESEARCH
The study of biologically relevant macromolecules at the
molecular level has been challenging for quite some time for
researchers. In recent years,
molecular modeling and
simulation
is the field of research that probably has more enthusiastically
contributed with atomistic information.
In computational studies, the
pH effects on
biologically relevant molecules have been addressed with several
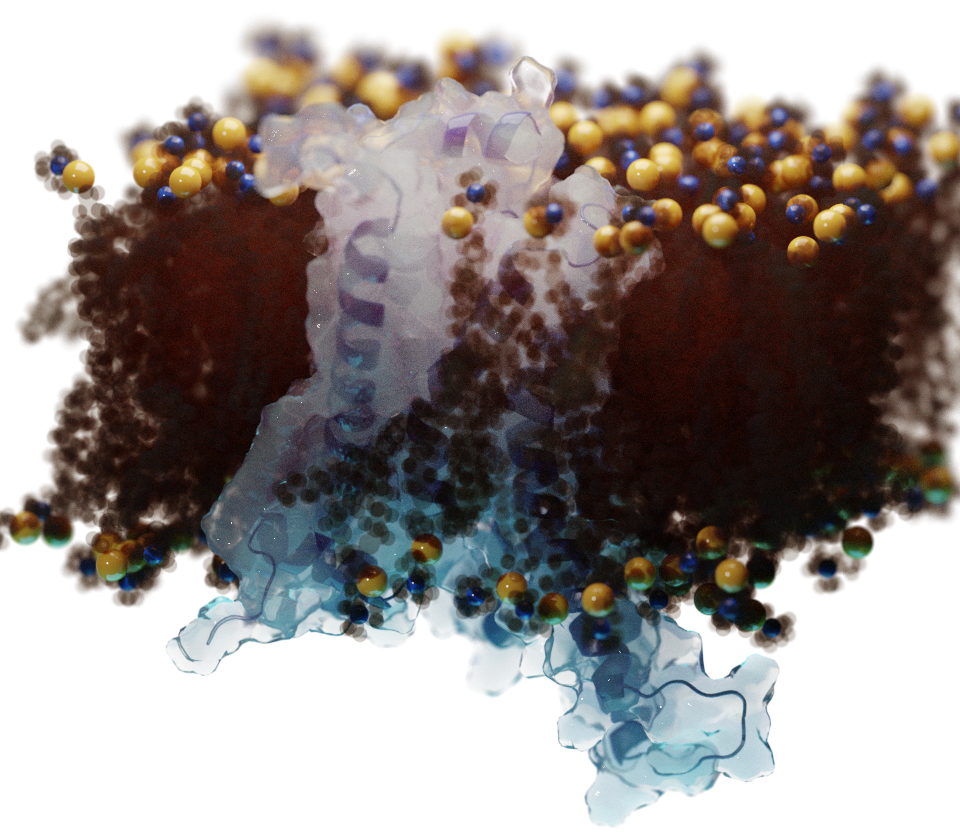
limitations. Biological membranes are inherently complex and
some important aspects, like protonation equilibrium, have also
been unsatisfactorily modeled. The complexity of the
membrane/water interface in biological membranes can even
increase many-fold due to the presence of a myriad of different
lipid molecules in their composition (e.g. anionic lipids). The
convoluted contribution of the complex electrostatic
interactions has rendered the problem of peptide, protein, drug,
and/or lipid protonation rather inaccessible, consequently
deterring their study. However, the gradual raise of
computational power, and the appearance of new and more refined
force-fields, have made it possible to model the water/membrane
region with increased realism.
The so-called
constant-pH MD (CpHMD)
methods aimed at solving the problem of including the pH effects
in MD simulations by allowing protonable groups to periodically
change their state during the simulation, thereby capturing the
coupling between conformation and protonation. In our group, we
are actively developing new CpHMD strategies that enable us to
study the dynamic properties of solutes (peptides, proteins, or
drugs) and lipid bilayers under different conditions of pH,
ionic strength, redox potential and solvent composition.